
It’s been about 8 years since I started working on my first 16S rRNA PCR survey (of Drosophila gut microbes). At that time, I was occasionally asked, “what about Archaea or what about microbial Eukayrotes?” Then, and ever since, my reply has been that it’s hard enough to get a handle on what’s going on with the bacteria – I don’t need to make my life more challenging by broadening my scope.
But, finally, this month, I’m making my life more challenging. As part of my new Seagrass Microbiome Project, I’ve decided to tackle the fungi. As far as I can tell, ITS is the “barcoding” marker of choice for fungal types. For many reasons, it’s best to follow the herd when doing this sort of thing: 1) someone else has already designed, tested, and published results with these primers, 2) there is a reasonably large database of ITS sequences available to compare my sequences to, and 3) I lack the interest and personnel to explore an alternative approach.
So, I just plunged right in. At first, I tried some new primers designed by Nick Bokulich, but he warned me that they were “finicky” and he was correct. I got no amplification with my seagrass samples, and the positive control I had only worked about half the time. I know some other fungi people, well, I know Jason Stajich (@hyphaltip), so I asked him which primers I should use, and I decided to go with the primers set used in a cool paper by Noah Fierer’s lab, in which they looked at fungi in rooftop gardens in New York City.
Those worked, and a few days ago I got word that my sequencing run was in. It looks like crap. We typically get about 12 million sequences from our MiSeq runs, but this time, I only got 4 million. I was also told that the reverse reads looked much better than the forward reads.
So, now, in addition to working with a new “barcode,” I have to troubleshoot a crappy sequencing run. In many ways, it’s nice to have undergrads and a technician in the lab who do all of my lab work for me these days, but it sucks when it’s time to troubleshoot because I’m so far removed from the bench that I have no idea what’s going on anymore.
So, the first thing I asked for was the Bioanalyzer trace that’s always run before the library goes on the machine. It looks like this:
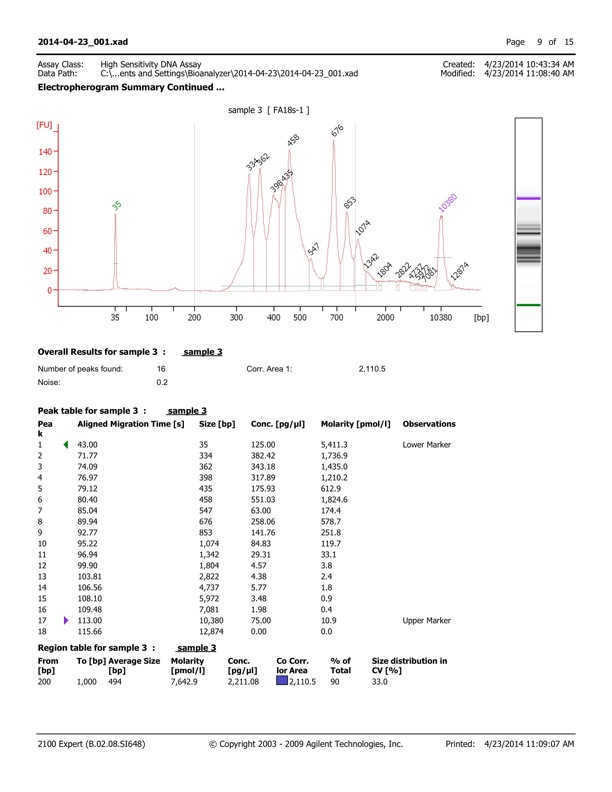
Bioanalyzer trace for my first fungal ITS MiSeq run
I had been told that there was size variation. I had even seen some of the PCR gels. But, still this is not what I expected to see. Upon seeing this, I am concerned about two things. 1) If there is strong preferential amplification of smaller DNA molecules during the bridge PCR on the flow cell, then will I even see DNA from those larger peaks? 2) With our 300bp reads, for sure the amplicons in the peaks <400 will have overlapping forward and reverse reads, but for sure the 676bp amplicons will not. What effect will these two things have on my analysis? How do I accommodate this size variation? One of the reasons to follow the herd with these methods is that other people have probably already encountered and dealt with exactly this issue, so I turned to Twitter…
There are some great resources suggested here. I know what I’ll be reading this weekend…

Pingback: Some notes on fungal ITS PCR surveys | The Seagrass Microbiome Project
Pingback: My foray into fungal ITS PCR surveys | Jonathan Eisen's Lab
Pingback: From Jenna Lang: What the fungi do I do with my ITS library? | microBEnet: The microbiology of the Built Environment network.
Pingback: Fungal ITS Taxonomy Problem: SOLVED (for now) | The Seagrass Microbiome Project